

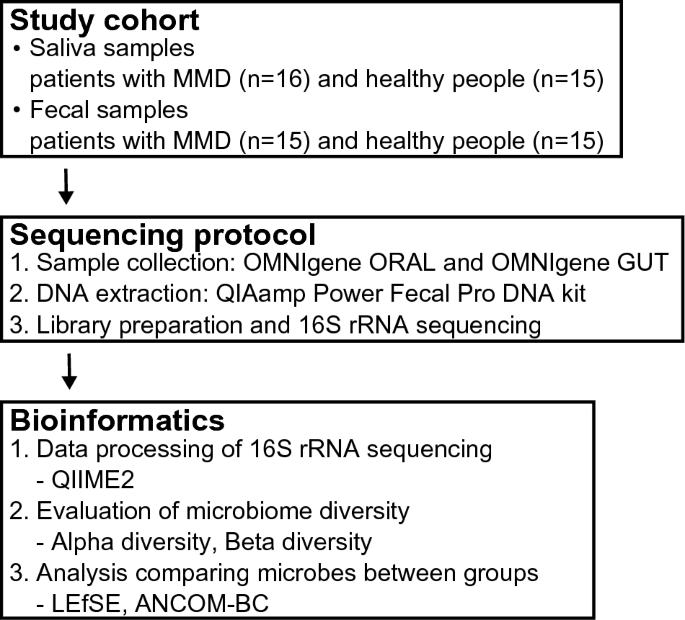
研究のフローチャートを図 3 に示します。
組織図を勉強します。
倫理の承認と参加者の同意
本研究は、名古屋大学医学部治験審査委員会により承認されました(承認番号 2022-0084、承認日 2022 年 6 月 3 日)。 すべての参加者または法的保護者から書面によるインフォームドコンセントを得ました。 この研究のすべての方法は、該当するガイドラインと規制に従って実行されました。
研究コホート
われわれは、2022年6月から2023年3月までに名古屋大学病院、地域医療機能推進機構中京病院、または豊田厚生病院で血行再建術を受けなかったMMD患者16人を前向きに募集した。 MMD の診断は、日本政府の厚生労働省によって承認された研究委員会によって提案されたガイドラインに基づいて行われました。21。 簡単に説明すると、診断基準は次のとおりです:(1)頭蓋内内頸動脈の末端部分の狭窄または閉塞、(2)脳底部のもやもや血管、および(3)血管造影上の特徴を伴う疾患の除外。 (例:自己免疫疾患、髄膜炎、脳腫瘍、ダウン症候群、神経線維腫症1型、または頭部の放射線照射後の脳血管病変)。 これらの患者は、抗生物質がマイクロバイオームに影響を与えたため、サンプル収集の前の 1 か月間は抗生物質を使用しませんでした。22。 16 人の MMD 患者から得られたすべてのサンプルのうち、1 つの糞便サンプルは収集が不十分だったために除外されました。 最後に、この研究には、MMD 患者からの 16 個の唾液サンプルと 15 個の糞便サンプルが含まれていました。
健康な対照群として、2022年6月から2023年3月まで名古屋大学で行われた研究に参加する意欲のある健康な人を募集した。健康な人は頭蓋手術を受けたことがなく、一過性脳虚血発作、虚血性脳卒中、頭蓋内脳卒中の病歴もなかった。 出血。 患者グループと同様に、健康な人はサンプル採取前の 1 か月間抗生物質を使用していませんでした。 収集した 17 サンプルの中から、年齢、性別、BMI の点で患者グループと一致する 15 サンプルを選択しました。
サンプル採取方法と微生物DNA抽出実験
OMNIgene ORAL および OMNIgene GUT (DNA Genotek、オタワ、カナダ) を使用して唾液および糞便サンプルを収集しました。 OMNIgene キットは、サンプル収集から 4 日以内に室温で良好なマイクロバイオーム安定性を実証しました23.24。 参加者はこれらのサンプルを自分で採取し、私たちは 4 日以内にそれらを収集しました。 収集直後、QIAamp Power Fecal Pro DNA キット (QIAGEN、ヒルデン、ドイツ) を製造元の指示に従って使用して、各サンプルから微生物 DNA を抽出しました。
ライブラリーの調製と 16S rRNA シーケンス
ライブラリーの調製は、16S メタゲノム シーケンシング ライブラリー調製プロトコル (Illumina、サンディエゴ、カリフォルニア州、米国) に従って実行されました。 微生物 16S rRNA 遺伝子の V3 ~ V4 領域は、KAPA HiFi HotStart ReadyMix (Kapa Bio) を使用して Amplicon 16S PCR フォワード プライマー (TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG) および Amplicon 16S PCR 逆プライマー (GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATTCTAATCC) を使用して増幅されました。システム)。 s、マサチューセッツ州ウィルミントン)25。 Illumine シーケンス アダプターとデュアル インデックス バーコードは、Nextera XT Index キット (Illumina) を使用して Amplicon 16S に追加されました。 ペアエンド 300 塩基対配列決定は、MiSeq v3 試薬キット (Illumina) を使用して Illumina MiSeq プラットフォームで実行されました。 ライブラリーの調製と配列決定はマクロジェン ジャパン株式会社によって行われました。
マイクロバイオームバイオインフォマティクス
16S rRNA シーケンス データの処理
16S rRNA シーケンスの結果は、Quantitative Insights into Microbial Ecology 2 (QIIME2) v2023.2 を使用して分析されました。26。 生のシーケンス データがインポートされ、逆多重化されました。 生成されたリードは、分割アンプリコンノイズ除去アルゴリズム 2 を使用してアンプリコン配列バリアント (ASV) にノイズ除去されました。27。 順方向読み取りと逆方向読み取りが減少し、品質スコア グラフの中央品質スコアが 20 を下回りました。28。 分類法は、Silva 138 99% 16S rRNA データベースに対してナイーブ ベイズ機械学習分類子 Scikit-learn を使用して ASV に割り当てられました。26.29。
マイクロバイオームの多様性の評価
アルファ多様性は、Chao1、観察された種、シャノンおよびシンプソン指数を計算することによって評価されました。 MMD 患者と健常者の間の統計的比較には、Mann-Whitney U 検定を使用し、重み付けされた UniFrac 距離と重み付けされていない UniFrac 距離を計算することによってベータ多様性を評価しました。 主座標分析 (PCoA) と順列多変量分散分析 (PERMANOVA) を実行しました。 これらの分析では、統計的有意性を p < 0.05 に設定しました。
MMD と対照群間の微生物を比較する分析: LEfSE と ANCOM-BC
グループ間で量が異なる微生物を特定するために、LEfSE 法と ANCOM-BC という 2 つの分析手法を属レベルまで適用しました。30.31。 分析方法間の結果の一致により、生物学的解釈の信頼性が向上します32。 LEfSe は、ノンパラメトリック統計検定を使用して微生物組成のパーセンテージを比較し、LDA スコアを計算して、異なる量で存在する各微生物の効果サイズを推定します。 ANCOM-BC は、未知のサンプリング割合を推定し、統計的テストの前にサンプル間の違いによって引き起こされるバイアスを補正しました。 この方法では、パラメトリック統計検定を使用して微生物組成の読み取り数を比較しました。 P 値は、Benjamini および Hochberg 法 (調整された p 値) を使用して調整され、調整された p 値 <0.05 を持つ属は、有意に差があり豊富な微生物であるとみなされました。33。
統計分析
参加者のベースライン特性を比較するために、マン・ホイットニーの U 検定を使用して数値データを比較し、ピアソンのカイ二乗検定またはフィッシャーの直接確率検定を使用してカテゴリデータを比較しました。 統計的有意性は p < 0.05 に設定されました。 統計分析は、R バージョン 4.2.2 (https://www.r-project.org/)。

「邪悪なポップカルチャーの擁護者。トラブルメーカー。不治のソーシャルメディアの魔術師。完全なインターネット愛好家。アマチュアのツイッター愛好家。流行に敏感な探検家。」





